Elimination, Clearance and Volume of Distribution¶
Pharmacokinetics is the study of “what the body does to the drug.” By and large, the body removes it either by metabolism (primarily in the liver) or by direct elimination of the unchanged drug (primarily in the kidneys).
Note
See the Elimination Example with KE parameter for the Tut Script used to generate results in this section.
Consider, as a first example, a bolus dose of 100 mg administered intravenously (IV) at time t0=1. Under first order kinetics, the rate of elimination is directly proportional to the amount of drug, $S(t)$, in the body at time t and the constant of proportionality is known as the elimination rate constant, KE:
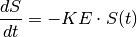
This ordinary differential equation (ODE) has a closed form solution:
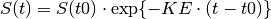
i.e. an exponential decay curve, where the initial conditions are
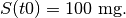
We can specify this directly in the PREDICTIONS section of the control script,
PREDICTIONS: |
plabel[DRUG_AMT] = "Drug Amount (mg)"
clabel[TIME] = "Time (minutes)"
p[DRUG_AMT] = c[AMT]*exp(-c[KE]*(c[TIME]-1.0))
c[DRUG_AMT] ~ norm(p[DRUG_AMT], 0.0)
where, for now, we treat KE as if it were a constant measurable quantity, c[KE], defined in the data file. From this closed form solution, we can predict the concentration at different time points and synthesize observations (Fig. 7).

Fig. 7 Amount Administered vs Time curve for an intravenously administered 100 mg bolus dose at time t0=1 with first order elimination. (Observations are noiseless in this example.)
Volume of Distribution¶
In practice, however, we cannot measure the amount of drug in the body from a blood plasma sample, only its concentration (i.e. amount of drug per unit volume).
Note
See the Elimination Example with Volume of Distribution for the Tut Script used to generate results in this section.
This measured concentration will clearly be influenced by the physiological volume of blood plasma in an individual’s body. Less obvious, however, is that the concentration will also be influenced by the physiochemical properties of the drug that determine how the drug is distributed throughout the body; some drugs are distributed mostly in the blood plasma (which is directly observed) whereas others get distributed to all tissues (which are not). The scaling factor – a combination of physiological and physiochemical properties – that relates amounts to concentrations is known as the volume of distribution, V, which varies greatly between drugs and, to a lesser extent, between individuals.
We model the observed concentration by including the volume of distribution parameter (with a value of 20 L in this example) in the PREDICTIONS section of the control script:
PREDICTIONS: |
plabel[DRUG_CONC] = "Drug Concentration (mg/L)"
clabel[TIME] = "Time (minutes)"
AMOUNT = c[AMT]*exp(-c[KE]*(c[TIME]-1.0))
p[DRUG_CONC] = AMOUNT/c[V]
c[DRUG_CONC] ~ norm(p[DRUG_CONC], 0.0)
which has the effect of scaling the observations (Fig. 8; note the scale of the y-axis).

Fig. 8 Concentration vs Time curve for an intravenously administered 100 mg bolus dose with first order elimination
Clearance¶
Note
See the Elimination Example with Clearance for the Tut Script used to generate results in this section.
Because we can only measure concentrations, it makes sense to define the rate of elimination also in terms of concentrations:
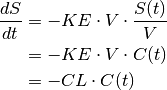
where
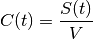
is the concentration at time t and
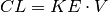
is a constant of proportionality known as the clearance, CL, that relates elimination to concentration. Again, this permits a closed form solution,
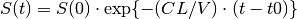
where we have substituted CL/V for the rate constant of elimination, KE:
PREDICTIONS: |
plabel[DRUG_CONC] = "Drug Concentration (mg/L)"
clabel[TIME] = "Time (minutes)"
AMOUNT = c[AMT]*exp(-(c[CL]/c[V])*(c[TIME]-1.0))
p[DRUG_CONC] = AMOUNT/c[V]
c[DRUG_CONC] ~ norm(p[DRUG_CONC], 0.0)
Because this is a mathematical equivalence, the Concentration vs Time curve (Fig. 8) is unchanged.